الرئيسية
أخبارعاجلة
رياضة
- الأخبار الرياضية
- أخبار الرياضة
- فيديو أخبار الرياضة
- نجوم الملاعب
- أخبار الرياضة
- ملاعب عربية وعالمية
- بطولات
- أخبار الأندية العربية
- مقابلات
- رياضة عربية
- رياضة عالمية
- موجب
- سالب
- مباريات ونتائج
- كرة الطائرة
- كرة اليد
- كرة السلة
- رمي
- قفز
- الجري
- تنس
- سيارات
- غولف
- سباق الخيل
- مصارعة
- جمباز
- أخبار المنتخبات
- تحقيقات
- مدونات
- أخبار المحترفين
- غاليري
ثقافة
إقتصاد
فن وموسيقى
أزياء
صحة وتغذية
سياحة وسفر
ديكور
آخر تحديث GMT22:15:33


آخر تحديث GMT22:15:33
اعتماد أول علاج جيني لاضطرابات الدم الموروثة

اضطرابات الدم
لندن - العرب اليوم
اعتمد منظمو الأدوية في المملكة المتحدة علاجاً جينياً يعدّ الأول من نوعه في العالم لاثنين من اضطرابات الدم الموروثة، حسب «استات نيوز» (Stat News). ويعدّ عقار «Casgevy» المخصص لمرض «الخلايا المنجلية» و«ثلاسيميا بيتا»، هو أول علاج يتم ترخيصه باستخدام أداة تعديل الجينات المعروفة باسم «كريسبر»، والتي حصل مخترعوها على جائزة نوبل في عام 2020.
وقالت «وكالة تنظيم الأدوية ومنتجات الرعاية الصحية البريطانية» إن العلاج مخصص للمرضى الذين تبلغ أعمارهم 12 عاماً أو أكثر «بعد تقييم دقيق لسلامته وجودته وفاعليته». ويذكر، أن مرض فقر الدم المنجلي و«ثلاسيميا بيتا» هي حالات وراثية ناجمة عن أخطاء في جينات الهيموغلوبين، والتي تستخدمها خلايا الدم الحمراء لنقل الأكسجين في جميع أنحاء الجسم. وينتشر مرض فقر الدم المنجلي بشكل خاص بين الأشخاص ذوي الأصول العائلية الأفريقية أو الكاريبية، بينما يصيب «ثلاسيميا بيتا» بشكل رئيسي الأشخاص من أصول دول البحر الأبيض المتوسط، وجنوب آسيا، وجنوب شرق آسيا والشرق الأوسط.
وتشمل أعراض مرض فقر الدم المنجلي ألماً شديداً وعدوى خطيرة ومهددة للحياة وفقر الدم. ويمكن أن يعاني الأشخاص المصابون بـ«الثلاسيميا بيتا» فقر دم حاداً، وغالباً ما يحتاج المرضى إلى نقل دم كل ثلاثة إلى خمسة أسابيع، إلى جانب الحقن والأدوية بصورة منتظمة.
وتم تصميم «Casgevy» للعمل عن طريق تعديل الجين المعيب في الخلايا الجذعية لنخاع العظم للمريض بحيث ينتج الجسم الهيموغلوبين الفعال. وفي التجربة السريرية لمرض الخلايا المنجلية، كان 28 من أصل 29 مريضاً في التحليل (97 في المائة) معافيين من أي ألم شديد طيلة 12 شهراً على الأقل بعد العلاج. وفي التجربة السريرية لـ«الثلاسيميا بيتا»، لم يكن 39 من أصل 42 مريضاً خضعوا للتحليل (93 في المائة) في حاجة إلى نقل خلايا الدم الحمراء لمدة 12 شهراً على الأقل بعد العلاج، في حين تراجعت حاجة المرضى الثلاثة الآخرين إلى الخضوع لعمليات نقل الخلايا الحمراء بواقع 70 في المائة.
قد يهمك أيضــــــــــــــــًا :
دراسة السمات الشخصية مرتبطة بخطر الاضطرابات الدماغية
- الإثنين ,24 شباط / فبراير GMT 18:25 2025 أطعمة تؤدي إلى تفاقم الالتهابات وتزيد من خطر الإصابة بالأمراض المزمنة




GMT 18:30 2025 الإثنين ,24 شباط / فبراير
فيروس كورونا جديد يثير القلق ويعيد مخاوف الأوبئة العالمية
GMT 18:25 2025 الإثنين ,24 شباط / فبراير
أطعمة تؤدي إلى تفاقم الالتهابات وتزيد من خطر الإصابة بالأمراض المزمنة
GMT 18:17 2025 الإثنين ,24 شباط / فبراير
5 دقائق يومياً من التمارين الخفيفة تحميك من الخرف
GMT 18:12 2025 الإثنين ,24 شباط / فبراير
5 عادات يومية مفاجئة تضر بالكبد دون أن تدرى
GMT 18:06 2025 الإثنين ,24 شباط / فبراير
أوقات يجب فيها تجنب ممارسة الرياضة
القاضية غادة عون تدّعي على رياض سلامة والحاكم الحالي لمصرف لبنان وآخرين

بيروت ـ العرب اليوم
ادعت النائب العام الاستئنافي في جبل لبنان القاضية غادة عون على كل من حاكم مصرف لبنان السابق رياض سلامة، وعلى الحاكم الحالي بالإنابة وسيم منصوري وعلى أنطوان سلامة ورجا أبو عسلة. وتمت التهم وفق مواد قانونية المتعلقة...المزيددينا الشربيني تواصل مفاجآتها في مسلسل "كامل العدد" ومحمد صلاح يدعمه مُجددًا وتكهنات حول ظهور ابنته مكة في الجزء الثالث

القاهرة ـ العرب اليوم
واصل صناع ونجوم مسلسل "كامل العدد ++" تشويق الجمهور لمفاجآت الجزء الثالث من المسلسل قبل أيام على انطلاق موسم العرض في رمضان 2025، وكشف نجوم المسلسل العديد من التفاصيل والمفاجآت الجديدة في العمل، وأثاروا التساؤلا...المزيدمنصة فيسبوك تفرض حداً زمنياً على الفيديوهات المباشرة

واشنطن ـ العرب اليوم
أعلنت شركة فيسبوك أن مقاطع الفيديو المباشرة ستظل متاحة على منصتها لمدة 30 يومًا فقط، وبعد ذلك سيتم حذفها تلقائيًا. وكان من الممكن سابقًا تخزين هذه المقاطع إلى أجل غير مسمى، لكن التغيير الجديد سيدخل حيز التنفيذ ال...المزيدعلماء يكتشفون ظاهرة كهرومغناطيسية غريبة داخل الهرم الأكبر في مصر

القاهرة ـ العرب اليوم
الأهرامات، تلك المعجزة التي أثارت الدهشة والحيرة عبر آلاف السنين، لا تزال تحتفظ بالعديد من الأسرار التي تحير العلماء، ومن بين هذه الأسرار، كشفت دراسة حديثة أن الهرم الأكبر في الجيزة في مصر والذي يعرف بهرم خوفو قد ي...المزيد


GMT 11:41 2025 الأحد ,23 شباط / فبراير
وزارة التربية والتعليم الفلسطينية تُعلن استئناف العام الدراسي في غزة للمرة الأولى منذ الحرب
GMT 12:05 2025 الإثنين ,24 شباط / فبراير
زيلينسكي يطالب بضمانات عسكرية مقابل اتفاقية المعادن المحتملة مع الولايات المتحدة
GMT 21:33 1970 الإثنين ,24 شباط / فبراير
بروكسل توافق على تعليق العقوبات على سوريا في مجال الطاقة والنفط والإنشاءات
GMT 16:05 2025 الإثنين ,24 شباط / فبراير
الصحف العالمية تحلل التشييع الجماهيري لنصر الله وصفي الدين ومستقبل حزب الله في لبنان
GMT 18:06 2025 الأحد ,23 شباط / فبراير
الجيش السوداني يعلن السيطرة على مدينتي الأبيض والقطينة
GMT 02:45 2025 الإثنين ,24 شباط / فبراير
الحكومة السودانية تنتقد اتفاق نيروبي واعتبر "حلما غير قابل للتنفيذ"
GMT 14:58 2025 الإثنين ,24 شباط / فبراير
منظمة الصحة العالمية تؤكد تقديم السعودية 500 مليون دولار لدعم جهود القضاء على شلل الأطفال حول العالم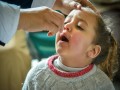
GMT 04:25 2025 الإثنين ,24 شباط / فبراير
قوات الاحتلال الاسرائيلي تقتحم بلدة سعير شمال الخليل
GMT 04:18 2025 الإثنين ,24 شباط / فبراير
«الصحة العالمية» تؤكد استهداف المرافق الصحية في الضفة انتهاكا للقانون الإنساني
GMT 04:28 2025 الإثنين ,24 شباط / فبراير
أستراليا تفرض غرامة مالية على تليغرام
GMT 04:20 2025 الإثنين ,24 شباط / فبراير
عقوبات جديدة ضد روسيا تفرضها أستراليا ونيوزيلندا
GMT 03:03 2025 الإثنين ,24 شباط / فبراير
تامر حسني يتكفل بمساعدة نجم الزمالك السابق إبراهيم الجمال "شيكا"
GMT 18:44 2025 الأحد ,23 شباط / فبراير
رانيا يوسف تكشف تفاصيل مغامرتها في رمضان مع "أهل الخطايا"
GMT 02:12 2025 الإثنين ,24 شباط / فبراير
ترامب يلغي 2000 وظيفة في الوكالة الأمريكية للتنمية الدولية
GMT 04:23 2025 الإثنين ,24 شباط / فبراير
هبوط آمن لرحلة أمريكان إيرلاينز في روما بعد تحويلها لأسباب أمنية
GMT 11:07 2025 الإثنين ,24 شباط / فبراير
جنازة حسن نصرالله
GMT 18:18 2025 الأحد ,23 شباط / فبراير
سبيس إكس تطلق 22 قمرا صناعيا جديدا لإنترنت ستارلينك
GMT 02:32 2025 الإثنين ,24 شباط / فبراير
الولايات المتحدة تسجل أعلى معدل وفيات بسبب السعال الديكي منذ عقود
GMT 12:25 2025 الأحد ,23 شباط / فبراير
الجيش السوداني يسيطر على النيل الأبيض واشتباكات ضارية في الخرطوم
GMT 01:30 2025 الإثنين ,24 شباط / فبراير
سماع دوي انفجارات في كييف وسط تحذير من ضربات جوية
GMT 00:41 2025 الإثنين ,24 شباط / فبراير
واتساب يحظر 8.4 مليون حساب في شهر واحد
GMT 02:23 2025 الإثنين ,24 شباط / فبراير
الحوثيون يستهدفون مقاتلة ومسيّرة أميركية بالصواريخ
Maintained and developed by Arabs Today Group SAL
جميع الحقوق محفوظة لمجموعة العرب اليوم الاعلامية 2025 ©
Maintained and developed by Arabs Today Group SAL
جميع الحقوق محفوظة لمجموعة العرب اليوم الاعلامية 2025 ©




أرسل تعليقك